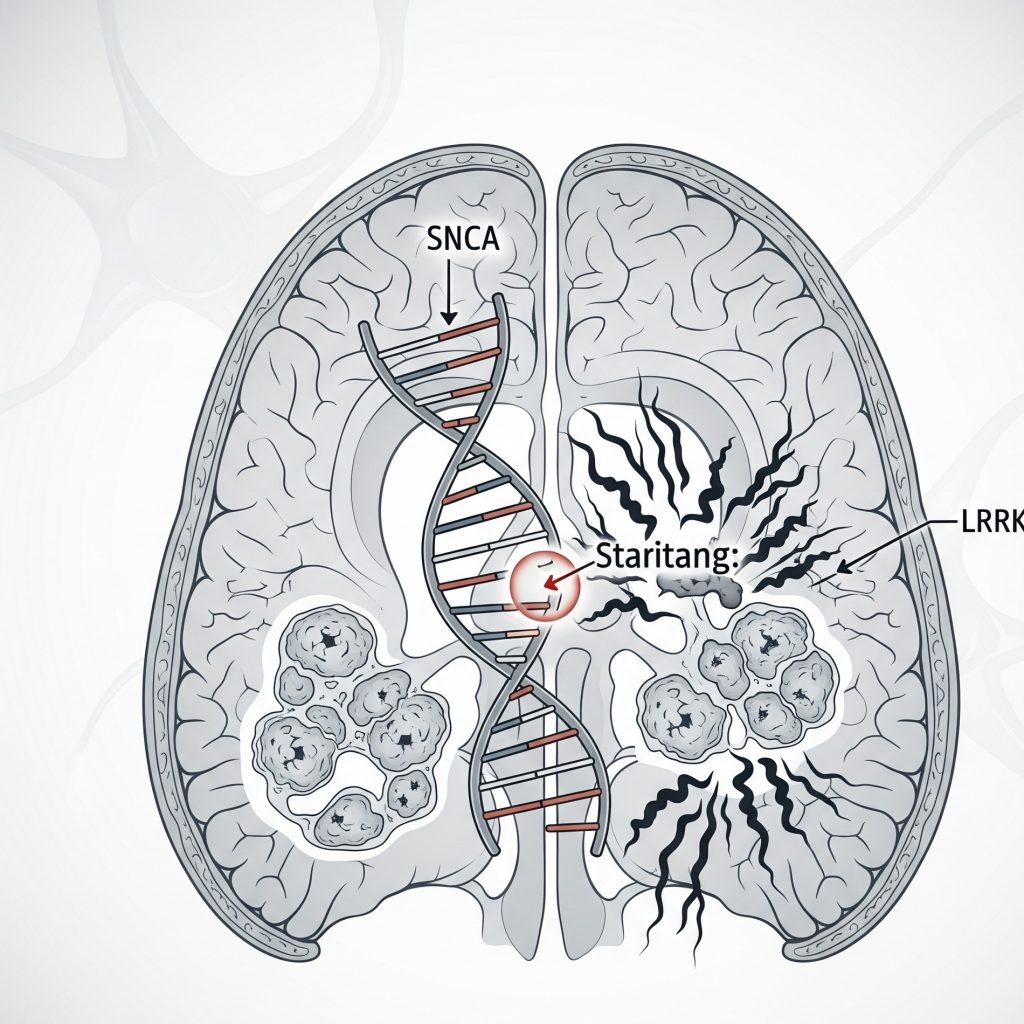
Генодиагностика болезни Паркинсона
Синонимы (rus): Болезнь Паркинсона
Синонимы (eng): Parkinson disease
Биоматериал: Венозная кровь
Показатель(и): Изменение копийности генов PARK1 (SNCA; alpha-synuclein), PARK2 (Parkin), PARK5 (UCHL1), PARK6 (PINK1), PARK7 (DJ1), PARK8 (Dardarin) и ATP13A2, две наиболее частые точковые мутации SNCA A30P, LRRK2 G2019S.
Метод(и): Полимеразная цепная реакция
Тип контейнера и особенности преаналитики: Пробирка для гематологических исследований с EDTA, 2 мл (фиолетовая крышечка)
Описание
Известно, что около 10-15% пациентов с болезнью Паркинсона имеют наследственный анамнез. На данный момент известно о 18 генах, мутации в которых могут вызывать развитие болезни Паркинсона. В комплексное генетическое исследование болезни Паркинсона входят маркеры на следующие формы заболевания: аутосомно-доминантные формы с мутациями в генах PARK1 (SNCA; точечная мутация A30P и делеции и дупликации гена ), PARK8 (LRRK2 мутация G2019S), аутосомно-рецессивные формы при гомозиготных мутациях в генах PARK2 (Parkin), PARK6 (PINK1), PARK7 (DJ1) и ATP13A2, мутации в гене PARK5 (UCHL1), ассоциированные с повышенным риском развития заболевания. Исследование рекомендуется пациентам с ранними формами болезни, атипичными проявлениями и резистентностью к проводимой терапии, а также при наличии семейного анамнеза заболевания.
Когда назначается
Дифференциальная диагностика атипичного паркинсонизма. Диагностика наследственных форм болезни Паркинсона.
Подготовка к анализу
Специальной подготовки не требуется. Исследование проводится натощак (не принимать пищу 3 часа до исследования, можно пить воду).
Расшифровка/Интерпретация
Отрицательный ответ исследования мутаций не исключает диагноз болезни Паркинсона. Гетерозиготная точечная мутация A30P и делеции и дупликации гена PARK1 (SNCA) вызывают раннюю форму болезни Паркинсона (появление первых симптомов до 50 лет) с первичным хорошим ответом на терапию леводопой, но быстрой прогрессией, ранним развитием деменции и когнитивной дисфункции, а также появлением атипичной симптоматики: центральной гиповентиляции и миоклонуса. Нужно отметить, что пенетрантность мутаций в гене PARK1 составляет 85%. Гомозиготные делеции и дупликации гена PARK2 (Parkin) вызывают аутосомно-рецессивную раннюю форму болезни Паркинсона (начало симптоматики в 30-40 лет). Кроме этого, при некоторых формах гетерозиготных мутаций и гомозиготных аберраций симптоматика может возникать в раннем детстве (ювенильная форма болезни Паркинсона). Обычно PARK2 –ассоциированная форма болезни Паркинсона характеризуется медленной прогрессией и отличным ответом на леводопу. Клинический фенотип пациентов с данной мутацией не отличим от случаев PINK1- и DJ-1- ассоциированной болезни Паркинсона. Точечные мутации и делеции и дупликации гена PARK5 (UCHL1) повышают риск развития болезни Паркинсона, но их патогенность до сих пор полностью не доказаны. Гомозиготные точечные мутации и делеции и дупликации гена PARK7 (DJ1) вызывают аутосомно-рецессивную раннюю форму болезни Паркинсона (начало симптоматики в 30-40 лет). Клинический фенотип пациентов с данной мутацией не отличим от случаев Parkin-и PINK1- ассоциированной болезни Паркинсона. Обычно PARK7 (DJ1) ассоциированная форма болезни Паркинсона характеризуется медленной прогрессией и отличным ответом на леводопу. Гетерозиготная мутация G2019S и делеции и дупликации гена PARK8 (LRRK2) вызывают позднюю форму болезни Паркинсона с медленной прогрессией. Пациенты хорошо отвечают на терапию леводопой и развитие деменции наблюдается редко. Гомозиготные точечные мутации и делеции и дупликации гена ATP13A2 вызывают аутосомно-рецессивную раннюю форму болезни Паркинсона (начало симптоматики в 30-40 лет). Кроме этого, мутации в данном гене вызывают атипичную форму болезни Паркинсона (Куфора-Ракеба), характеризующуюся ювенильной манифестацией, быстрой прогрессией, ассоциированной с деменцией, пирамидными знаками и надъядерным параличом взгляда. Гетерозиготная точечная мутация A30P и делеции и дупликации гена PARK1 (SNCA) вызывают раннюю форму болезни Паркинсона (появление первых симптомов до 50 лет) с первичным хорошим ответом на терапию леводопой, но быстрой прогрессией, ранним развитием деменции и когнитивной дисфункции, а также появлением атипичной симптоматики: центральной гиповентиляции и миоклонуса. Нужно отметить, что пенетрантность мутаций в гене PARK1 составляет 85%.
Список литературы
- Ahn TB, Kim SY, Kim JY, Park SS, Lee DS, Min HJ, Kim YK, Kim SE, Kim JM, Kim HJ, et al. 2008. α-Synuclein gene duplication is present in sporadic Parkinson disease. Neurology 70: 43–49 2. Camargos ST, Dornas LO, Momeni P, Lees A, Hardy J, Singleton A, Cardoso F 2009. Familial Parkinsonism and early onset Parkinson’s disease in a Brazilian movement disorders clinic: Phenotypic characterization and frequency of SNCA, PRKN, PINK1, and LRRK2 mutations. Mov Disord 24: 662–666